
As Joliot Curie was part of the urgent computing response action of GENCI and PRACE against the spread of COVID19 in 2020, this project is related with quantum chemistry and molecular dynamics (MD) aiming at developing new drugs. The extension of Joliot-Curie used for this project was acquired by GENCI through the PPI4HPC procurement process. The research was led by Luigi Genovese (CEA-Grenoble/Institut IRIG) and Michel Masella (CEA-Saclay / Institut Joliot).
The wavelet-based electronic structure code, BigDFT, implements Density Functional Theory calculations which scale linearly with the number of atoms. This contrasts with the traditional approaches which exhibit a cubic scaling behavior. Thanks to its unique features, full Quantum Mechanical descriptions of (solvated) proteins are accessible up to systems of tens of thousands of atoms.
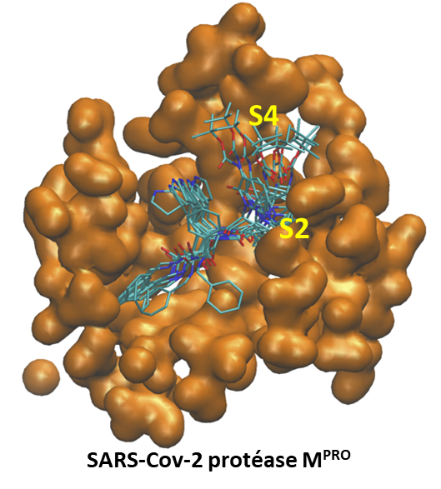
Combined with a molecular dynamics code, Polaris, BigDFT has been successfully applied to macro-molecular systems in biology, notably in connection with the COVID19 pandemic, by making it possible to identify the active fragments of a protein in the presence of a target molecule and to provide the key parameters for their modeling.
Targeting specific monoclonal antibodies can be routinely achieved, however increasing the antigen affinity as much as desired is still a challenging task. Most of the available theoretical tools in this field mainly focus on investigating close contact antibody/antigen (AA) local regions and usually ignore the effect on affinity of more distant domains. Because of the size of AA assemblies only standard pairwise molecular modeling force fields or empirical cost function are used to quantify the strength of their interactions. However, these theoretical approaches are known to be based on crude approximations making it impossible to achieve a level of accuracy which is sufficiently high.
This project, led by researchers from CEA, Joliot Institute and IRIG, and awarded a 2020 SANOFI iTech Award, adds two new steps to the standard computational protocols used to model AA assemblies (such as the popular Rosetta program package-based protocol) to evaluate and refine their solutions. It required the allocation of 15 million core hours on the AMD partition of Joliot Curie at TGCC. This extension was acquired by GENCI and installed at CEA through the PPI4HPC project delivering record-breaking performance and enabling more powerful supercomputing systems needed for such demanding operations.

Figure: Fragment analysis of a molecule with indication of activated fragments based on a Jupyter notebook in Python for post-processing automation
The two additional steps of the standard computational protocols involve:
(1) investigating the potential energy surface of AA from "Replica Exchange" molecular dynamics simulations based on a polarizable multiscale molecular modeling approach
(2) refining the simulation results using the BigDFT complexity reduction framework, which allows the calculation of the quantum interaction energy of complete AA assemblies.
Find more information is available here: https://chemrxiv.org/engage/chemrxiv/article-details/60c75006ee301c7adac7a7d5
